Ab initio
Istilah “Ab initio” adalah bahasa latin untuk “dari awal”. Nama ini diberikan kepada perhitungan yang berasal langsung dari prinsip-prinsip teoritis, tanpa masuknya data eksperimen. Sebagian besar saat ini adalah mengacu ke perhitungan perkiraan kuantum mekanik. Perkiraan yang dibuat biasanya perkiraan matematika, seperti menggunakan bentuk fungsional sederhana untuk fungsi atau mendapatkan solusi perkiraan untuk sebuah persamaan diferensial.
Jenis yang paling umum perhitungan ab initio ini disebut perhitungan Hartree Fock (disingkat HF), di mana pendekatan utama disebut pendekatan lapangan pusat. Ini berarti bahwa tolakan Coulomb elektron-elektron tidak secara khusus diperhitungkan. Namun, itu efek bersih adalah termasuk dalam perhitungan. Ini adalah variasi perhitungan, yang berarti bahwa energi perkiraan dihitung semua sama atau lebih besar daripada energi yang tepat. Energi dihitung biasanya dalam satuan yang disebut Hartrees (1 H = 27,2114 eV). Karena pendekatan lapangan pusat, energi dari perhitungan HF selalu lebih besar daripada energi yang tepat dan cenderung ke nilai membatasi disebut batas Hartree Fock.
Pendekatan kedua dalam perhitungan HF adalah bahwa fungsi gelombang harus dijelaskan oleh beberapa bentuk fungsional, yang hanya diketahui secara tepat untuk satu beberapa sistem elektron. Fungsi yang paling sering digunakan adalah kombinasi linier dari orbital tipe Slater exp (-ax) atau jenis orbital Gaussian exp (-ax ^ 2), disingkat STO dan GTO. Fungsi gelombang terbentuk dari kombinasi linier dari orbital atom atau lebih sering dari kombinasi linear dari fungsi dasar. Karena pendekatan ini, perhitungan HF paling memberikan energi dihitung lebih besar dari batas Fock Hartree. Himpunan tepat fungsi dasar yang digunakan sering ditentukan oleh singkatan, seperti STO-3G atau 6-311 g + + **.
Sejumlah jenis perhitungan dimulai dengan perhitungan HF kemudian benar untuk tolakan elektron-elektron eksplisit, disebut sebagai korelasi. Beberapa metode ini Mohlar-Plesset teori perturbasi (MPN, dimana n adalah urutan koreksi), yang Generalized Valence Bond (GVB) metode, Multi-Konfigurasi Self Konsisten Lapangan (MCSCF), Interaksi konfigurasi (CI) dan teori Cluster Ditambah (CC). Sebagai kelompok, metode ini disebut sebagai perhitungan berkorelasi.
Sebuah metode, yang menghindari membuat kesalahan HF di tempat pertama disebut Quantum Monte Carlo (QMC). Ada beberapa rasa QMC .. variasional, difusi dan fungsi Green. Metode-metode ini bekerja dengan fungsi gelombang dan mengevaluasi secara eksplisit berkorelasi integral numerik menggunakan integrasi Monte Carlo. Perhitungan ini bisa sangat memakan waktu, tetapi mereka mungkin metode yang paling akurat dikenal saat ini.
Metode ab initio adalah alternatif teori kerapatan fungsional (DFT), di mana total energi dinyatakan dalam total kepadatan elektron, bukan fungsi gelombang. Dalam jenis ini perhitungan, ada Hamilton dan perkiraan ekspresi perkiraan untuk kepadatan total elektron.
Sisi baik metode ab initio adalah bahwa mereka akhirnya bertemu dengan solusi yang tepat, setelah semua perkiraan yang dibuat cukup kecil di besarnya. Namun, konvergensi ini tidak montonic. Kadang-kadang, perhitungan terkecil memberikan hasil yang terbaik untuk properti tertentu.
Sisi buruk dari metode ab initio adalah bahwa mereka mahal. Metode ini sering mengambil sejumlah besar waktu komputer cpu, memori dan ruang disk. Metode skala HF sebagai N 4, dimana N merupakan jumlah fungsi dasar, sehingga perhitungan dua kali lebih besar membutuhkan 16 kali lebih lama untuk menyelesaikan. perhitungan Korelasi sering skala jauh lebih buruk dari ini. Dalam prakteknya, solusi sangat akurat hanya dapat diperoleh ketika molekul berisi setengah lusin elektron atau kurang.
Secara umum, perhitungan ab initio kualitatif memberikan hasil yang sangat baik dan dapat memberikan hasil kuantitatif semakin akurat sebagai molekul yang dimaksud menjadi lebih kecil.
Semiempirical
Semiempirical perhitungan ditetapkan dengan struktur umum yang sama sebagai perhitungan HF. Dalam kerangka ini, potongan informasi tertentu, seperti dua integral elektron, yang didekati atau sama sekali dihilangkan. Dalam rangka untuk mengoreksi kesalahan diperkenalkan dengan menghilangkan bagian dari perhitungan, metode ini parameter, dengan melakukan suaian kurva dalam beberapa parameter atau nomor, untuk memberikan kesepakatan yang terbaik dengan data eksperimen.
Sisi baik dari perhitungan semiempirical adalah bahwa mereka jauh lebih cepat daripada perhitungan ab initio.
Sisi buruk dari perhitungan semiempirical adalah bahwa hasilnya bisa tidak menentu. Jika molekul yang sedang dihitung mirip dengan molekul dalam basis data yang digunakan untuk parameterisasi metode, maka hasilnya mungkin akan sangat baik. Jika molekul yang dihitung secara signifikan berbeda dari apa pun di set parameterisasi, jawaban mungkin sangat miskin.
perhitungan Semiempirical telah sangat sukses dalam deskripsi kimia organik, di mana hanya ada beberapa elemen digunakan secara luas dan molekul yang ukuran sedang. Namun, metode semiempirical telah dirancang khusus untuk deskripsi kimia anorganik juga.
Pemodelan solid state
Struktur elektronik dari kristal tak terbatas didefinisikan oleh struktur plot band, yang memberikan energi orbital elektron untuk setiap titik di k-ruang, yang disebut zona Brillouin. Sejak ab initio dan perhitungan semiempirical hasil energi orbital, mereka dapat diterapkan untuk band perhitungan struktur. Namun, jika memakan waktu untuk menghitung energi untuk molekul, itu bahkan lebih memakan waktu untuk menghitung energi untuk daftar poin di zona Brillouin.
perhitungan struktur Band telah dilakukan untuk sistem yang sangat rumit, namun perangkat lunak belum cukup otomatis atau cukup cepat sehingga siapa pun tidak struktur band santai. Jika Anda ingin melakukan perhitungan struktur band, Anda sebaiknya berharap untuk menempatkan banyak waktu dalam usaha Anda.
Mekanika molekul
Jika molekul terlalu besar untuk secara efektif menggunakan pengobatan semiempirical, masih mungkin untuk model perilaku itu dengan menghindari mekanika kuantum benar-benar. Metode disebut sebagai mekanika molekul membentuk ekspresi aljabar sederhana untuk energi total senyawa, tanpa keharusan untuk menghitung fungsi gelombang atau kepadatan total elektron. Ekspresi energi terdiri dari persamaan klasik sederhana, seperti persamaan osilator harmonik dalam rangka untuk menggambarkan energi yang berkaitan dengan ikatan peregangan, membungkuk, rotasi dan gaya antarmolekul, seperti interaksi van der Waals dan ikatan hidrogen. Semua konstanta dalam persamaan ini harus diperoleh dari data percobaan atau perhitungan ab initio.
Dalam metode mekanika molekul, basis data senyawa yang digunakan untuk parameterisasi metode (satu set parameter dan fungsi yang disebut medan gaya) sangat penting untuk keberhasilan itu. Dimana sebagai metode semiempirical mungkin parameter terhadap satu set molekul organik, sebuah metode mekanika molekul mungkin parameter terhadap kelas khusus molekul, seperti protein. Medan gaya seperti ini hanya akan diharapkan memiliki relevansi untuk menjelaskan protein lain.
Sisi baik dari mekanika molekuler adalah bahwa hal itu memungkinkan pemodelan molekul besar, seperti protein dan segmen dari DNA, sehingga alat utama ahli biokimia komputasi.
Sisi buruk dari mekanika molekul adalah bahwa ada banyak sifat-sifat kimia yang bahkan tidak didefinisikan dalam metode ini, seperti keadaan tereksitasi elektronik. Dalam rangka untuk bekerja dengan sistem yang sangat besar dan rumit, sering molekul mekanik paket perangkat lunak yang paling kuat dan paling mudah untuk menggunakan antarmuka grafis. Karena itu, mekanik kadang-kadang digunakan karena mudah, tetapi belum tentu cara yang baik untuk menjelaskan sistem.
Dinamika molekul
dinamika molekul terdiri dari memeriksa perilaku tergantung waktu dari molekul, seperti gerak getaran atau gerak Brown. Hal ini paling sering dilakukan dalam sebuah tulisan mekanik klasik mirip dengan perhitungan mekanika molekul.
Penerapan dinamika molekuler untuk pelarut / sistem terlarut memungkinkan perhitungan properti seperti koefisien difusi atau fungsi distribusi radial untuk digunakan dalam perawatan mekanik statistik. Biasanya skema perhitungan pelarut / zat terlarut adalah bahwa jumlah molekul (mungkin 1000) diberikan beberapa posisi awal dan kecepatan. posisi baru menghitung waktu kecil kemudian berdasarkan gerakan ini dan proses ini itterated untuk ribuan langkah untuk membawa sistem untuk keseimbangan dan memberikan gambaran statistik yang baik dari fungsi distribusi radial.
Dalam rangka untuk menganalisa getaran molekul tunggal, banyak dinamika langkah-langkah yang dilakukan, maka data tersebut Fourier berubah menjadi domain frekuensi. Sebuah puncak yang diberikan dapat dipilih dan diubah kembali ke domain waktu, untuk melihat apa gerakan pada frekuensi yang terlihat seperti.
Statistik Mekanika
Mekanika statistika adalah matematika berarti mengekstrapolasi sifat termodinamika bahan curah dari deskripsi molekul material. Banyak mekanika statistik masih pada tahap kertas dan pensil teori, karena mekanika kuantum tidak dapat menyelesaikan persamaan Schrödinger tepat lagi, mekanika statistik tidak benar-benar memiliki bahkan titik awal yang baik untuk perlakuan yang benar-benar ketat. Mekanika statistika perhitungan sering ditempelkan ke akhir perhitungan inito ab untuk properti fasa gas. Untuk properti fasa terkondensasi, sering molekul dinamika perhitungan diperlukan dalam rangka untuk melakukan percobaan komputasi.
Termodinamika
Termodinamika adalah salah satu deskripsi paling baik dikembangkan kimia matematika. Sangat sering setiap pengobatan termodinamika yang tersisa untuk pena dan kertas kerja sepele karena banyak aspek kimia begitu akurat digambarkan dengan ekspresi matematika yang sangat sederhana.
Struktur-Properti Hubungan
Struktur-properti hubungan yang kualitatif atau kuantitatif didefinisikan secara empiris hubungan antara struktur molekul dan sifat diamati. Dalam beberapa kasus ini mungkin tampak duplikat hasil mekanik statistik, namun sistem struktur-properti hubungan tidak perlu didasarkan pada prinsip-prinsip teoritis ketat.
Kasus yang paling sederhana hubungan struktur-properti aturan jempol kualitatif. Misalnya, kimia polimer yang berpengalaman mungkin dapat memprediksi apakah polimer akan halus atau rapuh berdasarkan geometri dan ikatan monomer.
Ketika struktur-properti hubungan yang disebutkan dalam literatur saat ini, biasanya menyiratkan hubungan matematis kuantitatif. Hubungan ini paling sering diperoleh dengan menggunakan software curve fitting untuk menemukan kombinasi linear dari sifat molekul, yang paling mereproduksi properti yang diinginkan. Sifat-sifat molekul biasanya diperoleh dari perhitungan pemodelan molekul. deskriptor molekul lain seperti berat molekul atau deskripsi topologi juga digunakan.
Ketika properti yang dijelaskan adalah properti fisik, seperti titik didih, ini disebut sebagai Kuantitatif Struktur-Properti Relationship (QSPR). Ketika properti yang dijelaskan adalah jenis aktivitas biologis (seperti aktivitas obat), ini disebut sebagai Kuantitatif Struktur-Aktivitas Relationship (HKSA).
Perhitungan Simbolik
perhitungan simbolik dilakukan bila sistem yang terlalu besar untuk sebuah deskripsi atom-by-atom masih layak pada setiap tingkat pendekatan. Sebuah contoh mungkin gambaran membran dengan menjelaskan lipid individu sebagai perwakilan beberapa poligon dengan beberapa ekspresi untuk energi interaksi. Pengobatan semacam ini digunakan untuk biokimia komputasi dan bahkan mikrobiologi.
Kecerdasan Buatan
Teknik diciptakan oleh ilmuwan komputer tertarik dalam kecerdasan buatan telah diterapkan sebagian besar berupa rancangan obat dalam beberapa tahun terakhir. Metode ini juga pergi dengan nama De Novo atau desain obat rasional. Skenario umum adalah bahwa beberapa situs fungsional telah diidentifikasi dan diinginkan untuk datang dengan struktur molekul yang akan berinteraksi dengan situs bahwa untuk menghalangi fungsi itu. Daripada memiliki seorang ahli kimia mencoba ratusan atau ribuan kemungkinan dengan program mekanika molekul, mekanika molekul dibangun ke dalam program kecerdasan buatan, yang mencoba jumlah besar “masuk akal” kemungkinan dalam fasion otomatis. Jumlah teknik untuk menggambarkan “cerdas” bagian dari operasi ini begitu beragam yang tidak mungkin untuk membuat generalisasi tentang bagaimana hal ini diimplementasikan dalam program.
Bagaimana melakukan proyek riset komputasi
Bila menggunakan kimia komputasi untuk menjawab pertanyaan kimia, masalah jelas adalah bahwa Anda perlu tahu bagaimana menggunakan perangkat lunak. Permasalahan yang terjawab adalah bahwa Anda perlu untuk mengetahui seberapa baik jawabannya akan menjadi. Berikut adalah daftar periksa untuk diikuti.
Apa yang Anda ingin tahu? Seberapa akurat? Mengapa? Jika Anda tidak dapat menjawab pertanyaan-pertanyaan, maka Anda bahkan tidak memiliki proyek penelitian belum.
Seberapa akurat Anda memprediksi jawabannya akan? Dalam kimia analitik, Anda melakukan sejumlah pengukuran identik kemudian bekerja keluar kesalahan dari deviasi standar. Dengan percobaan komputasi, melakukan hal yang sama harus selalu memberikan hasil yang sama persis. Cara yang Anda memperkirakan kesalahan Anda adalah untuk membandingkan sejumlah perhitungan mirip dengan jawaban eksperimental. Ada artikel dan kompilasi dari studi ini. Jika tidak ada, Anda akan perlu menebak metode mana yang harus masuk akal, didasarkan pada asumsi itu kemudian melakukan penelitian sendiri, sebelum Anda dapat menerapkannya pada Anda tidak diketahui dan punya ide seberapa bagus perhitungannya. Ketika seseorang hanya memberitahu Anda dari atas kepala mereka metode apa yang digunakan, mereka juga memiliki jumlah wajar dari jenis informasi hafal, atau mereka tidak tahu apa yang mereka bicarakan. Waspadalah terhadap seseorang yang memberitahu Anda sebuah program yang diberikan adalah baik hanya karena itu adalah satu-satunya mereka tahu bagaimana menggunakan, bukan mendasarkan jawaban mereka pada kualitas hasil.
Berapa lama Anda berharap untuk mengambil? Jika dunia yang sempurna, Anda akan memberitahu PC Anda (suara masukan tentu saja) untuk memberikan solusi yang tepat untuk persamaan Schrödinger dan melanjutkan hidup Anda. Namun, sering kali perhitungan ab initio akan memakan sehingga waktu yang dibutuhkan waktu satu dekade untuk melakukan perhitungan tunggal, jika Anda bahkan memiliki mesin dengan cukup memori dan ruang disk. Namun, sejumlah metode yang ada karena setiap yang terbaik untuk situasi tertentu. Caranya adalah dengan menentukan mana yang terbaik untuk proyek Anda. Sekali lagi, jawabannya adalah untuk melihat ke dalam literatur dan melihat berapa lama masing-masing diperlukan. Jika satu-satunya yang Anda tahu adalah bagaimana skala perhitungan, melakukan perhitungan sederhana yang mungkin kemudian gunakan persamaan skala untuk memperkirakan berapa lama waktu yang diperlukan untuk melakukan semacam perhitungan bahwa Anda telah diprediksi akan memberikan akurasi yang diinginkan.
Apa perkiraan sedang dilakukan? Yang signifikan? Ini adalah bagaimana Anda menghindari tampak seperti orang bodoh yang lengkap, ketika anda berhasil melakukan perhitungan yang sampah lengkap. Sebuah contoh akan mencoba untuk mencari tahu tentang gerak getaran yang sangat anharmonic, ketika perhitungan menggunakan pendekatan osilator harmonik.
Setelah Anda akhirnya menjawab semua pertanyaan ini, Anda siap untuk benar-benar melakukan perhitungan. Sekarang Anda harus menentukan software apa yang tersedia, berapa biayanya dan bagaimana menggunakannya. Perhatikan bahwa dua program dari jenis yang sama (ab initio yaitu) dapat menghitung sifat-sifat yang berbeda, sehingga Anda harus memastikan program ini tidak persis apa yang Anda inginkan.
Ketika Anda sedang belajar bagaimana menggunakan sebuah program, Anda dapat mencoba untuk melakukan puluhan perhitungan yang akan gagal karena Anda dibangun masukan salah. Jangan gunakan molekul proyek Anda untuk melakukan hal ini. Membuat semua kesalahan Anda dengan sesuatu yang sangat mudah, seperti molekul air. Dengan begitu Anda tidak membuang sejumlah besar waktu.
Visualisasi
visualisasi data adalah proses menampilkan informasi dalam jenis representasi piktorial atau grafis. Sejumlah program komputer yang sekarang tersedia untuk menerapkan skema pewarnaan data atau bekerja dengan tiga dimensi representasi.

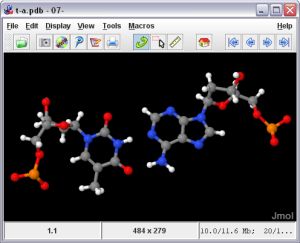 Jmol ini gartis, merupakan penampil strukutur molekul tiga dimensi (molecule viewer) yang dapat digukan secara bebas oleh siapapun yang menekuni bidang kimia dan biokimia. Aplikasi ini merupakan cross-platform, berjalan di sistem operasi Windows, Mac OS X, dan Linux / Unix. Fitur yang dimilikinya di antaranya membaca berbagai jenis file dan output dari program kimia kuantum, dan animasi file multi-frame. JmolApplet adalah applet web browser yang dapat diintegrasikan ke dalam halaman situs. Aplikasi Jmol adalah aplikasi Java standalone yang berjalan di desktop. JmolViewer merupakan seperangkat alat yang dapat diintegrasikan ke dalam aplikasi Java lainnya.
Jmol ini gartis, merupakan penampil strukutur molekul tiga dimensi (molecule viewer) yang dapat digukan secara bebas oleh siapapun yang menekuni bidang kimia dan biokimia. Aplikasi ini merupakan cross-platform, berjalan di sistem operasi Windows, Mac OS X, dan Linux / Unix. Fitur yang dimilikinya di antaranya membaca berbagai jenis file dan output dari program kimia kuantum, dan animasi file multi-frame. JmolApplet adalah applet web browser yang dapat diintegrasikan ke dalam halaman situs. Aplikasi Jmol adalah aplikasi Java standalone yang berjalan di desktop. JmolViewer merupakan seperangkat alat yang dapat diintegrasikan ke dalam aplikasi Java lainnya.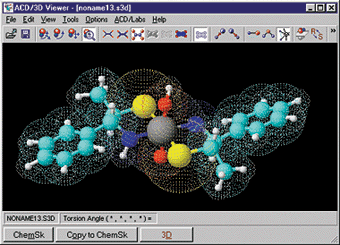



 Banyak hal yang bisa dimanfaatkan dari MarvinSketch untuk pengajaran kimia. Gambar dan animasi molekul dapat dibuat dengan mudah. Selanjutnya dapat disisipkan dalam media pembelajaran kimia. Tutorial berupa video untuk mengefektifkan MarvinSkecth dapat dilihat atau
Banyak hal yang bisa dimanfaatkan dari MarvinSketch untuk pengajaran kimia. Gambar dan animasi molekul dapat dibuat dengan mudah. Selanjutnya dapat disisipkan dalam media pembelajaran kimia. Tutorial berupa video untuk mengefektifkan MarvinSkecth dapat dilihat atau